Introduction
Depuis qu’elle a été décrite pour la première fois en association avec la polyarthrite rhumatoïde1, la dermatite granulomateuse interstitielle (DGI) a été associée à des maladies hématologiques2-4, en particulier aux syndromes myélodysplasiques, qui représentent la plupart des cas. Nous rapportons ici le premier cas de dermatite granulomateuse non interstitielle (DGNI) associée à une polycythémie vraie (PV) et un nouveau cas de DGI associée à une myélodysplasie.
Descriptions des casPatient 1
La patiente était une femme espagnole de 72 ans qui n’avait jamais voyagé en dehors de l’Europe et qui avait des antécédents de PV traitée par hydroxyurée et acide acétylsalicylique. Elle a signalé l’apparition de lésions cutanées diffuses 5 ans après le diagnostic de sa maladie hématologique. Les lésions consistaient en 15 à 20 plaques érythémateuses mesurant 1 à 3 cm sur le dos, la poitrine et la surface extérieure des deux bras. Les diagnostics proposés étaient une infection, une dermatose neutrophilique et un lupus tumidus (Fig. 1). La biopsie initiale a révélé un infiltrat inflammatoire lymphocytaire superficiel et profond autour des vaisseaux, des follicules et des annexes, sans dépôt de mucine. Les résultats étaient compatibles avec un diagnostic de lupus tumidus. Le patient a refusé le traitement par hydroxychloroquine. Les lésions ont fini par disparaître sans traitement, bien qu’elles soient réapparues. Dix-huit mois plus tard, des lésions similaires aux lésions d’origine et 4 gros nodules érythémateux indolores sont apparus sur les deux jambes, indiquant une panniculite lupique. L’analyse d’un nouveau prélèvement biopsique a montré un infiltrat lymphocytaire périvasculaire superficiel et profond composé de cellules matures et sans atypie cytologique (CD4+, CD2+ et CD5+ ; CD8+, CD79+ et CD20+ ; occasionnellement CD30+) accompagné d’histiocytes (CD68+) et de plasmocytes. Aucune anomalie épidermique ou hypodermique ni mucine n’ont été observées. L’hydroxyurée et l’acide acétylsalicylique ont été suspendus malgré le faible degré de suspicion de causalité, et un traitement par doxycycline 100mg/j a été prescrit pendant 2 mois, bien que les lésions n’aient pas complètement disparu. Un an plus tard, le patient a connu une nouvelle poussée de nodules et de plaques sur les membres supérieurs et inférieurs. Les poils du corps, la sensibilité et la transpiration n’étaient pas affectés (Fig. 2). L’examen de la biopsie a révélé un infiltrat inflammatoire histiocytaire superficiel et profond qui tendait à former des granulomes interstitiels dans certaines zones et des granulomes nodulaires dans d’autres (sans nécrose fibrinoïde). Les granulomes étaient entourés de lymphocytes matures (CD4+, CD8-, CD20-, et CD30-) sans atypie cytologique (Fig. 3). Aucun corps histiocytaire intracytoplasmique évocateur de leishmaniose n’a été observé. De même, aucun matériel exogène n’était visible à l’œil nu ou en lumière polarisée. Les résultats de la coloration de Ziehl-Neelsen, de la culture (bactéries, mycobactéries et champignons) et des tests sérologiques (virus de l’immunodéficience humaine et syphilis) étaient négatifs. Les résultats des réactifs de phase aiguë et des études d’auto-immunité (anticorps antinucléaires, anticorps anti-ADN natif, anticorps anti-antigène nucléaire extractible et complément) étaient normaux. L’amplification en chaîne par polymérase n’a pas été réalisée pour exclure la leishmaniose, car les lésions intermittentes se sont résorbées avec des corticostéroïdes ou sans traitement. Une fois le diagnostic de dermatite granulomateuse confirmé, les médicaments précédemment interrompus ont été réintroduits, sans que l’état du patient ne s’aggrave. A l’heure actuelle, le patient est stable sous prednisone toutes les 48 heures et, si l’infiltration a diminué, elle n’a pas disparu.
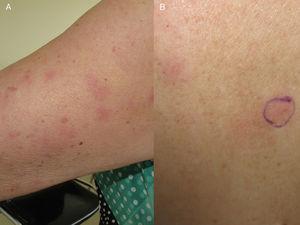
A, plaques érythémateuses sur la face externe du bras. B, lésions similaires sur le dos.
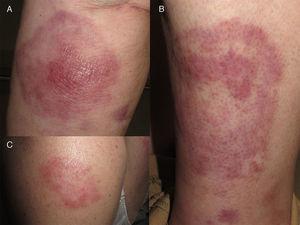
A, Plaque érythémateuse ronde avec infiltration marquée au centre du nodule. B, plaque érythémateuse annulaire sur l’autre jambe. C, plaque isolée sur le bras.
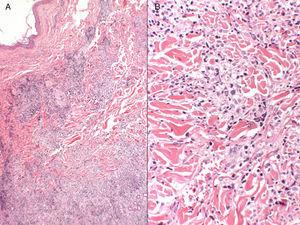
A, Infiltrat inflammatoire dermique superficiel et profond (hématoxyline-éosine, grossissement original, ×4). B, Tendance à former des granulomes interstitiels dans certaines zones, avec un aspect plus nodulaire dans d’autres (pas de nécrose fibrinoïde), entourés de lymphocytes matures sans atypies cytologiques (hématoxyline-éosine, grossissement original, ×10).
Patient 2
Le patient était un homme espagnol de 74 ans qui n’avait jamais voyagé hors d’Espagne. Il avait été diagnostiqué avec une anémie réfractaire avec excès de blastes de type 1 un an auparavant et a été référé avec des lésions cutanées accompagnées d’arthralgie et aucun signe d’arthrite franche. Les lésions étaient des plaques érythémateuses mesurant 2 à 3 cm sur le front, le cou et les joues. La sensibilité, la pilosité et la transpiration n’étaient pas affectées (Fig. 4). Trois mois plus tôt, il avait présenté des lésions similaires un mois après le premier cycle de traitement par azacytidine ; les lésions ont complètement disparu sous prednisone à 0,5mg/kg/jour. La biopsie cutanée a révélé un infiltrat inflammatoire périannexiel superficiel et profond constitué de lymphocytes matures sans atypie accompagnés de granulomes histiocytaires interstitiels (sans nécrose) (Fig. 5), de rares éosinophiles et de cellules géantes multinucléées. Une dégénérescence vacuolaire de la membrane basale et des kératinocytes nécrotiques ont été observés dans des zones spécifiques. La microscopie standard et la microscopie en lumière polarisée n’ont révélé aucun corps étranger ou corps intracytoplasmique. Les résultats des cultures et des tests sérologiques pour la syphilis et le virus de l’immunodéficience humaine étaient négatifs. L’immunohistochimie (coloration CD15) a révélé la présence de quelques cellules. Les résultats de l’immunofluorescence directe (IgG, IgM, IgA, C3 et fibrinogène) et de la coloration de Ziehl-Neelsen étaient également négatifs. Le bilan sanguin était remarquable – hormis la myélodysplasie du patient – pour les taux de protéine C-réactive (16,7mg/dL) et la vitesse de sédimentation des érythrocytes (88mm). Tous les autres paramètres – anticorps antinucléaires, anticorps anti-ADN natif, anticorps anti-antigène nucléaire extractible et complément (C3 et C4) – étaient normaux. Comme dans le cas précédent, aucune étude moléculaire visant à exclure la leishmaniose n’a été réalisée. Les lésions ont disparu sans traitement au bout d’un mois et ne sont pas réapparues avec les cycles ultérieurs d’azacytidine. Trois ans plus tard, la myélodysplasie a évolué vers une leucémie myéloïde aiguë.
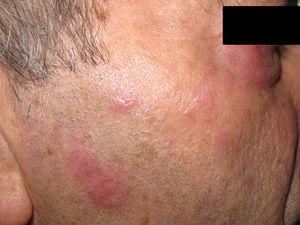
Plaques érythémateuses sur la joue et la paupière inférieure.
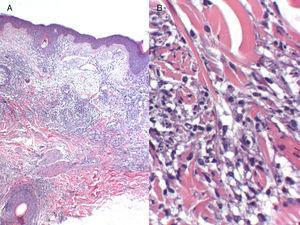
A, Infiltrat inflammatoire périvasculaire et périannexiel superficiel et profond (hématoxyline-éosine, grossissement original, ×4). B, Infiltrat inflammatoire composé de lymphocytes matures et de granulomes histiocytaires interstitiels, sans nécrose (hématoxyline-éosine, grossissement original, ×20).
Discussion
En 1993, Vestey et al.5 ont rapporté les 2 premiers cas de lésions cutanées granulomateuses associées au syndrome myélodysplasique ; il s’agissait d’éruptions papulaires diffuses dont l’histologie évoquait une sarcoïdose dans un cas et un granulome annulaire dans l’autre. Toujours en 1993, Ackerman1 a rapporté pour la première fois le profil de l’IGD, qui dans ce cas était associé à la polyarthrite rhumatoïde, comme un infiltrat dermique composé principalement d’histiocytes répartis de manière interstitielle et en palissades autour de petits faisceaux de collagène dégénéré, avec peu ou pas de dépôts de mucine et un nombre limité d’éosinophiles et de neutrophiles. Katz6 a été le premier à décrire une éruption granulomateuse cutanée qui différait du granulome annulaire, de la sarcoïdose et de l’IGD. L’éruption a été décrite comme un infiltrat histiocytaire multinodulaire, périvasculaire et périannexiel avec de petits lymphocytes et plasmocytes. Comme chez le patient 2 (voir ci-dessus), cette éruption impliquait des nodules érythémateux de taille moyenne (1-5cm) sur le visage, le cuir chevelu et le cou. L’auteur a décrit l’observation comme une simple éruption cutanée granulomateuse. Le rapport a été publié en raison de l’association entre l’éruption et la myélodysplasie. Aung et al.7 ont récemment utilisé le terme NIGD pour décrire un cas associé à la myélodysplasie ; le cas rapporté par Katz pourrait être inclus sous ce terme. Le rapport de cas décrit une grande plaque érythémateuse couvrant le bras du patient et imitant une cellulite, suivie de l’apparition de 2 plaques linéaires sur le coude. Cornejo et al.8 ont effectué une analyse conjointe de spécimens de biopsies d’IGD et de NIGD à différents stades de développement des lésions d’un même patient atteint de myélodysplasie ; le premier correspondait à des lésions papuleuses et le second à des nodules. Il convient de noter que, dans ce cas, un troisième spécimen de biopsie provenant d’une phase plus disséminée de la maladie cutanée du patient a révélé que le motif granulomateux se confondait avec la leucémie cutis (cellules à gros noyaux hyperchromatiques positifs à la myéloperoxydase). La progression de la leucémie a été confirmée par la suite dans la moelle osseuse.
En plus des résultats cliniques d’une éruption papulaire, de nodules et d’une grande plaque mimant une pyodermatite rapportés à ce jour, il convient d’attirer l’attention sur les résultats de Patsinakidis et al,9 qui ont signalé de grandes plaques urticariformes et annulaires sur les cuisses, le tronc et les bras, avec une IGD prouvée par biopsie dans le contexte de la myélodysplasie. La maladie la plus étendue a été rapportée par Balin et al,10 qui ont décrit une IGD confirmée par histopathologie sous forme de plaques et de papules coalescentes sur le tronc et les membres et couvrant 80 % de la surface corporelle totale.
Dans le premier cas que nous rapportons, les spécimens de biopsie ont révélé divers schémas histopathologiques se développant en parallèle, démontrant ainsi un spectre histologique au sein de ce processus réactif.
Les cas que nous rapportons ici sont le premier cas où le NIGD est associé à la PV et le septième où la dermatite granulomateuse est associée à la myélodysplasie dans un contexte large (granulome annulaire, sarcoïdose, IGD et NIGD). Nous pensons que ces cas devraient être regroupés sous le terme plus large de dermatite granulomateuse, qui inclurait tous les sous-types, privilégiant ainsi le fait que ces granulomes sont une manifestation réactive ou paranéoplasique de troubles hématologiques, tels que la myélodysplasie et la PV. Par conséquent, au moins 1 hémogramme complet devrait être réalisé dans le cadre de l’étude d’extension chez ces patients.
Divulgations éthiquesProtection de l’homme et de l’animal
Les auteurs déclarent qu’aucun test n’a été effectué sur l’homme ou l’animal dans le cadre de cette étude.
Confidentialité des données
Les auteurs déclarent qu’aucune donnée privée de patient n’apparaît dans cet article.
Droit à la vie privée et au consentement éclairé.